Analysing 20k cells
2025-03-01
Last updated: 2025-03-01
Checks: 7 0
Knit directory: muse/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200712) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 6550d2d. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Ignored: analysis/figure/
Ignored: data/1M_neurons_filtered_gene_bc_matrices_h5.h5
Ignored: data/293t/
Ignored: data/293t_3t3_filtered_gene_bc_matrices.tar.gz
Ignored: data/293t_filtered_gene_bc_matrices.tar.gz
Ignored: data/5k_Human_Donor1_PBMC_3p_gem-x_5k_Human_Donor1_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/5k_Human_Donor2_PBMC_3p_gem-x_5k_Human_Donor2_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/5k_Human_Donor3_PBMC_3p_gem-x_5k_Human_Donor3_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/5k_Human_Donor4_PBMC_3p_gem-x_5k_Human_Donor4_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/Parent_SC3v3_Human_Glioblastoma_filtered_feature_bc_matrix.tar.gz
Ignored: data/brain_counts/
Ignored: data/cl.obo
Ignored: data/cl.owl
Ignored: data/jurkat/
Ignored: data/jurkat:293t_50:50_filtered_gene_bc_matrices.tar.gz
Ignored: data/jurkat_293t/
Ignored: data/jurkat_filtered_gene_bc_matrices.tar.gz
Ignored: data/pbmc20k/
Ignored: data/pbmc20k_seurat/
Ignored: data/pbmc3k/
Ignored: data/pbmc4k_filtered_gene_bc_matrices.tar.gz
Ignored: data/refdata-gex-GRCh38-2020-A.tar.gz
Ignored: data/seurat_1m_neuron.rds
Ignored: data/t_3k_filtered_gene_bc_matrices.tar.gz
Ignored: r_packages_4.4.1/
Untracked files:
Untracked: analysis/bioc_scrnaseq.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/pbmc20k.Rmd) and HTML
(docs/pbmc20k.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 6550d2d | Dave Tang | 2025-03-01 | Use BPCells matrices from the start |
| html | 607fc5d | Dave Tang | 2025-02-24 | Build site. |
| Rmd | 6daf5ce | Dave Tang | 2025-02-24 | Saving and loading on-disk backed Seurat objects |
| html | 4dc6978 | Dave Tang | 2025-02-23 | Build site. |
| Rmd | 9e472aa | Dave Tang | 2025-02-23 | Convert to sparse matrix for annotation |
| html | ffad125 | Dave Tang | 2025-02-23 | Build site. |
| Rmd | 0e6715c | Dave Tang | 2025-02-23 | Annotate using SingleR |
| html | 3a0e769 | Dave Tang | 2025-02-23 | Build site. |
| Rmd | aad6fa2 | Dave Tang | 2025-02-23 | Normalised and scaled data are stored as iterable matrices |
| html | 9a02923 | Dave Tang | 2025-02-22 | Build site. |
| Rmd | 6c0d4f0 | Dave Tang | 2025-02-22 | Convert sparse matrix to iterable matrix |
| html | f6301ec | Dave Tang | 2025-02-22 | Build site. |
| Rmd | 22af89e | Dave Tang | 2025-02-22 | Analysing 20k cells |
Data
Read HDF5 file paths into a list.
hdf5_files <- list.files(path = "data", pattern = "5k_Human", full.names = TRUE)
hdf5_files[1] "data/5k_Human_Donor1_PBMC_3p_gem-x_5k_Human_Donor1_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5"
[2] "data/5k_Human_Donor2_PBMC_3p_gem-x_5k_Human_Donor2_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5"
[3] "data/5k_Human_Donor3_PBMC_3p_gem-x_5k_Human_Donor3_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5"
[4] "data/5k_Human_Donor4_PBMC_3p_gem-x_5k_Human_Donor4_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5"Read raw counts into a list of matrices.
mats <- purrr::map(seq_along(hdf5_files), function(x){
my_mat <- BPCells::open_matrix_10x_hdf5(hdf5_files[x])
colnames(my_mat) <- paste0('donor', x, '_', colnames(my_mat))
my_mat
})
str(mats, max.level = 1)List of 4
$ :Formal class 'RenameDims' [package "BPCells"] with 4 slots
$ :Formal class 'RenameDims' [package "BPCells"] with 4 slots
$ :Formal class 'RenameDims' [package "BPCells"] with 4 slots
$ :Formal class 'RenameDims' [package "BPCells"] with 4 slotsGet gene names.
mat_tmp <- Seurat::Read10X_h5(hdf5_files[1])
my_genes <- row.names(mat_tmp)
rm(mat_tmp)
gc() used (Mb) gc trigger (Mb) max used (Mb)
Ncells 10995803 587.3 16559960 884.4 14635520 781.7
Vcells 19679453 150.2 85287993 650.7 106593458 813.3Combine the matrices.
combined_matrix <- do.call(cbind, mats)
row.names(combined_matrix) <- my_genes
combined_matrix38606 x 22203 IterableMatrix object with class RenameDims
Row names: DDX11L2, MIR1302-2HG ... ENSG00000277196
Col names: donor1_AAACCAAAGGTGACGA-1, donor1_AAACCCTGTGACGAGT-1 ... donor4_TGTGTTGAGTTACGGC-1
Data type: uint32_t
Storage order: column major
Queued Operations:
1. Concatenate cols of 4 matrix objects with classes: RenameDims, RenameDims ... RenameDims (threads=0)
2. Reset dimnamesSeurat object
Create Seurat object from the list of matrices.
pbmc20k <- CreateSeuratObject(
counts = combined_matrix,
min.cells = 3,
min.features = 200
)
pbmc20kAn object of class Seurat
29157 features across 22061 samples within 1 assay
Active assay: RNA (29157 features, 0 variable features)
1 layer present: countsCounts layer.
pbmc20k@assays$RNA$counts29157 x 22061 IterableMatrix object with class RenameDims
Row names: DDX11L2, ENSG00000238009 ... ENSG00000278817
Col names: donor1_AAACCAAAGGTGACGA-1, donor1_AAACCCTGTGACGAGT-1 ... donor4_TGTGTTGAGTTACGGC-1
Data type: uint32_t
Storage order: column major
Queued Operations:
1. Concatenate cols of 4 matrix objects with classes: RenameDims, RenameDims ... RenameDims (threads=0)
2. Reset dimnamesDonor information in orig.ident.
head(pbmc20k@meta.data) orig.ident nCount_RNA nFeature_RNA
donor1_AAACCAAAGGTGACGA-1 donor1 42841 7087
donor1_AAACCCTGTGACGAGT-1 donor1 4896 2104
donor1_AAACGAATCAGGCTAC-1 donor1 12498 3564
donor1_AAACGACAGATTGACT-1 donor1 22198 4371
donor1_AAACGATGTCTTGAAC-1 donor1 10305 2945
donor1_AAACGATGTGCGCGAA-1 donor1 15948 4161Use {BPCells} to store the matrix in the Seurat object to on-disk matrices. Note, that this is only possible for V5 assays.
BPCells::write_matrix_dir(
mat = pbmc20k@assays$RNA$counts,
dir = 'data/pbmc20k',
overwrite = TRUE
)29157 x 22061 IterableMatrix object with class MatrixDir
Row names: DDX11L2, ENSG00000238009 ... ENSG00000278817
Col names: donor1_AAACCAAAGGTGACGA-1, donor1_AAACCCTGTGACGAGT-1 ... donor4_TGTGTTGAGTTACGGC-1
Data type: uint32_t
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc20kpbmc20k.mat <- open_matrix_dir(dir = "data/pbmc20k")
pbmc20k@assays$RNA$counts <- pbmc20k.mat
pbmc20k@assays$RNA$counts29157 x 22061 IterableMatrix object with class RenameDims
Row names: DDX11L2, ENSG00000238009 ... ENSG00000278817
Col names: donor1_AAACCAAAGGTGACGA-1, donor1_AAACCCTGTGACGAGT-1 ... donor4_TGTGTTGAGTTACGGC-1
Data type: uint32_t
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc20k
2. Reset dimnamesSeurat workflow
Process with the Seurat 4 workflow.
options(future.globals.maxSize = 2 * 1024^3)
fixed_PrepDR5 <- function(object, features = NULL, layer = 'scale.data', verbose = TRUE) {
layer <- layer[1L]
olayer <- layer
layer <- SeuratObject::Layers(object = object, search = layer)
if (is.null(layer)) {
abort(paste0("No layer matching pattern '", olayer, "' not found. Please run ScaleData and retry"))
}
data.use <- SeuratObject::LayerData(object = object, layer = layer)
features <- features %||% VariableFeatures(object = object)
if (!length(x = features)) {
stop("No variable features, run FindVariableFeatures() or provide a vector of features", call. = FALSE)
}
if (is(data.use, "IterableMatrix")) {
features.var <- BPCells::matrix_stats(matrix=data.use, row_stats="variance")$row_stats["variance",]
} else {
features.var <- apply(X = data.use, MARGIN = 1L, FUN = var)
}
features.keep <- features[features.var > 0]
if (!length(x = features.keep)) {
stop("None of the requested features have any variance", call. = FALSE)
} else if (length(x = features.keep) < length(x = features)) {
exclude <- setdiff(x = features, y = features.keep)
if (isTRUE(x = verbose)) {
warning(
"The following ",
length(x = exclude),
" features requested have zero variance; running reduction without them: ",
paste(exclude, collapse = ', '),
call. = FALSE,
immediate. = TRUE
)
}
}
features <- features.keep
features <- features[!is.na(x = features)]
features.use <- features[features %in% rownames(data.use)]
if(!isTRUE(all.equal(features, features.use))) {
missing_features <- setdiff(features, features.use)
if(length(missing_features) > 0) {
warning_message <- paste("The following features were not available: ",
paste(missing_features, collapse = ", "),
".", sep = "")
warning(warning_message, immediate. = TRUE)
}
}
data.use <- data.use[features.use, ]
return(data.use)
}
assignInNamespace('PrepDR5', fixed_PrepDR5, 'Seurat')
seurat_wf_v4 <- function(seurat_obj, scale_factor = 1e4, num_features = 2000, num_pcs = 30, cluster_res = 0.5, debug_flag = FALSE){
seurat_obj <- NormalizeData(seurat_obj, normalization.method = "LogNormalize", scale.factor = scale_factor, verbose = debug_flag)
seurat_obj <- FindVariableFeatures(seurat_obj, selection.method = 'vst', nfeatures = num_features, verbose = debug_flag)
seurat_obj <- ScaleData(seurat_obj, verbose = debug_flag)
seurat_obj <- RunPCA(seurat_obj, verbose = debug_flag)
seurat_obj <- RunHarmony(seurat_obj, "orig.ident")
seurat_obj <- RunUMAP(seurat_obj, reduction = "harmony", dims = 1:num_pcs, verbose = debug_flag)
seurat_obj
}
pbmc20k <- seurat_wf_v4(pbmc20k)Transposing data matrixInitializing state using k-means centroids initializationHarmony 1/10Harmony 2/10Harmony 3/10Harmony converged after 3 iterationsWarning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
This message will be shown once per sessionNormalised and scaled data are stored as IterableMatrix
objects.
pbmc20k@assays$RNA$data29157 x 22061 IterableMatrix object with class RenameDims
Row names: DDX11L2, ENSG00000238009 ... ENSG00000278817
Col names: donor1_AAACCAAAGGTGACGA-1, donor1_AAACCCTGTGACGAGT-1 ... donor4_TGTGTTGAGTTACGGC-1
Data type: double
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc20k
2. Reset dimnames
3. Convert type from uint32_t to double
4. Scale by 1e+04
5. Scale columns by 2.33e-05, 0.000204 ... 7.7e-05
6. Transform log1p
7. Reset dimnamespbmc20k@assays$RNA$scale.data2000 x 22061 IterableMatrix object with class RenameDims
Row names: HES4, ISG15 ... ENSG00000275063
Col names: donor1_AAACCAAAGGTGACGA-1, donor1_AAACCCTGTGACGAGT-1 ... donor4_TGTGTTGAGTTACGGC-1
Data type: double
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc20k
2. Select rows: 24, 25 ... 29156 and cols: all
3. Reset dimnames
4. Convert type from uint32_t to double
5. Scale by 1e+04
6. Scale columns by 2.33e-05, 0.000204 ... 7.7e-05
7. Transform log1p
8. Select rows: 497, 1853 ... 446 and cols: all
9. Reset dimnames
10. Transform min by row: 2.8, 2.49 ... 0.38
11. Scale rows by 3.59, 4.04 ... 26.4
12. Shift rows by -0.0687, -0.0609 ... -0.049
13. Select rows: 1252, 443 ... 604 and cols: all
14. Reset dimnamesUMAP.
DimPlot(pbmc20k, reduction = "umap", group.by = "orig.ident", pt.size = .1)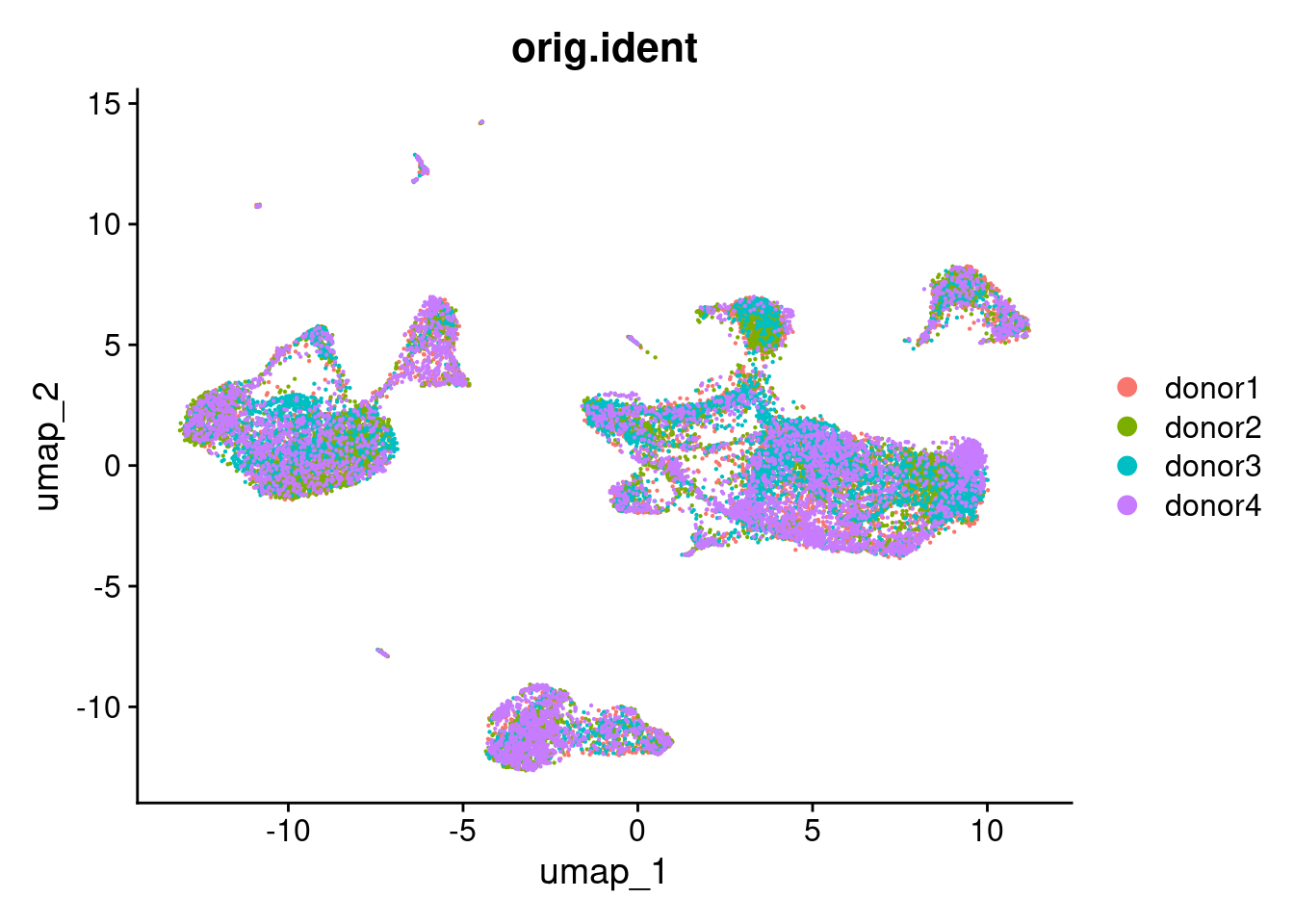
| Version | Author | Date |
|---|---|---|
| f6301ec | Dave Tang | 2025-02-22 |
Annotation
Annotate using {SingleR}.
monaco_immune <- fetchReference("monaco_immune", "2024-02-26")
monaco_immuneclass: SummarizedExperiment
dim: 46077 114
metadata(0):
assays(1): logcounts
rownames(46077): A1BG A1BG-AS1 ... ZYX ZZEF1
rowData names(0):
colnames(114): DZQV_CD8_naive DZQV_CD8_CM ... G4YW_Neutrophils
G4YW_Basophils
colData names(3): label.main label.fine label.ontpbmc20k.anno <- SingleR(
test=as(pbmc20k@assays$RNA$data, "sparseMatrix"),
ref=monaco_immune,
labels=colData(monaco_immune)$label.main
)Warning: Converting to a dense matrix may use excessive memory
This message is displayed once every 8 hours.Warning in asMethod(object): sparse->dense coercion: allocating vector of size
4.8 GiBhead(pbmc20k.anno)DataFrame with 6 rows and 4 columns
scores labels
<matrix> <character>
donor1_AAACCAAAGGTGACGA-1 0.320884:0.260316:0.480567:... T cells
donor1_AAACCCTGTGACGAGT-1 0.150790:0.128674:0.302692:... CD4+ T cells
donor1_AAACGAATCAGGCTAC-1 0.250532:0.239444:0.435132:... CD4+ T cells
donor1_AAACGACAGATTGACT-1 0.318373:0.340789:0.126101:... Monocytes
donor1_AAACGATGTCTTGAAC-1 0.228895:0.202186:0.417512:... CD4+ T cells
donor1_AAACGATGTGCGCGAA-1 0.467819:0.245565:0.253680:... B cells
delta.next pruned.labels
<numeric> <character>
donor1_AAACCAAAGGTGACGA-1 0.0813905 T cells
donor1_AAACCCTGTGACGAGT-1 0.0978551 CD4+ T cells
donor1_AAACGAATCAGGCTAC-1 0.0634572 CD4+ T cells
donor1_AAACGACAGATTGACT-1 0.1820548 Monocytes
donor1_AAACGATGTCTTGAAC-1 0.0908428 CD4+ T cells
donor1_AAACGATGTGCGCGAA-1 0.1411577 B cellsAdd annotations to metadata.
cbind(
pbmc20k@meta.data,
as.data.frame(pbmc20k.anno)
) -> pbmc20k@meta.dataUMAP with annotations.
DimPlot(pbmc20k, reduction = "umap", group.by = "labels", pt.size = .1, label = TRUE, repel = TRUE)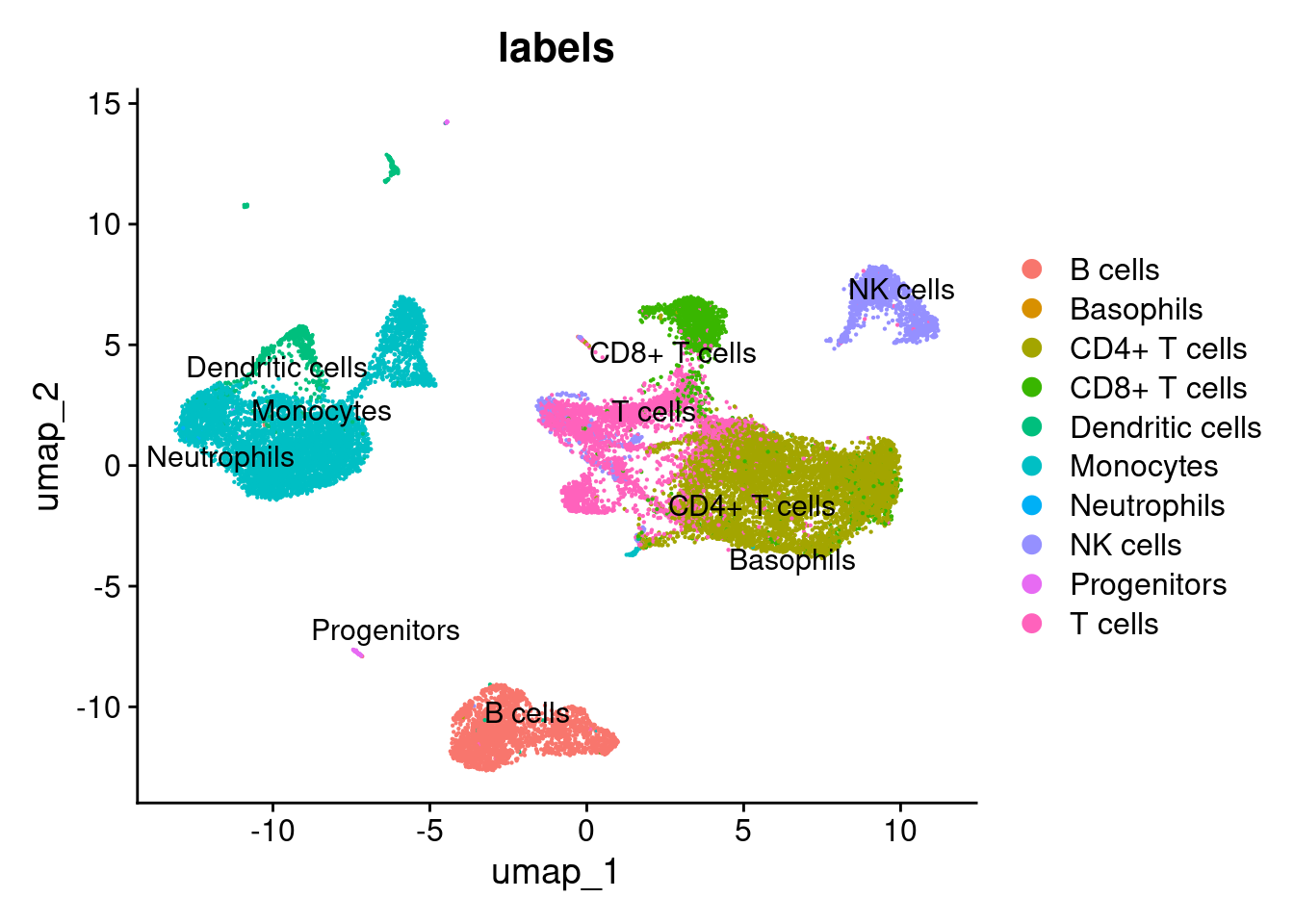
| Version | Author | Date |
|---|---|---|
| ffad125 | Dave Tang | 2025-02-23 |
Saving and loading
If you save your object and load it in in the future, Seurat will
access the on-disk matrices by their path, which is stored in the assay
level data. To make it easy to ensure these are saved in the same place,
we provide new functionality to the SaveSeuratRds()
function. In this function, you specify your filename. The pointer to
the path in the Seurat object will change to the current directory.
This also makes it easy to share your Seurat objects with BPCells matrices by sharing a folder that contains both the object and the BPCells directory.
Make sure you use a different directory than where the on-disk matrices are stored or they will be recursively copied.
output_dir <- "data/pbmc20k_seurat"
if(!dir.exists(output_dir)){
dir.create(output_dir)
}
SaveSeuratRds(
object = pbmc20k,
file = paste0(output_dir, "/pbmc20k.rds")
)Warning: Trying to move '/home/rstudio/muse/data/pbmc20k' to itself, skipping
Trying to move '/home/rstudio/muse/data/pbmc20k' to itself, skipping
Trying to move '/home/rstudio/muse/data/pbmc20k' to itself, skippinglist.files(output_dir)[1] "pbmc20k" "pbmc20k.rds"list.files(paste0(output_dir, "/pbmc20k")) [1] "col_names" "idxptr" "index_data"
[4] "index_idx" "index_idx_offsets" "index_starts"
[7] "row_names" "shape" "storage_order"
[10] "val_data" "val_idx" "val_idx_offsets"
[13] "version" Need to use LoadSeuratRds() to load or else none of the
layers will be imported.
pbmc20k_import <- LoadSeuratRds(paste0(output_dir, '/pbmc20k.rds'))
pbmc20k_importAn object of class Seurat
29157 features across 22061 samples within 1 assay
Active assay: RNA (29157 features, 2000 variable features)
3 layers present: counts, data, scale.data
3 dimensional reductions calculated: pca, harmony, umappbmc20k_import@assays$RNA$counts29157 x 22061 IterableMatrix object with class RenameDims
Row names: DDX11L2, ENSG00000238009 ... ENSG00000278817
Col names: donor1_AAACCAAAGGTGACGA-1, donor1_AAACCCTGTGACGAGT-1 ... donor4_TGTGTTGAGTTACGGC-1
Data type: uint32_t
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc20k
2. Reset dimnames
sessionInfo()R version 4.4.1 (2024-06-14)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 22.04.5 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] celldex_1.16.0 SingleR_2.8.0
[3] SummarizedExperiment_1.36.0 Biobase_2.66.0
[5] GenomicRanges_1.58.0 GenomeInfoDb_1.42.3
[7] IRanges_2.40.1 S4Vectors_0.44.0
[9] BiocGenerics_0.52.0 MatrixGenerics_1.18.1
[11] matrixStats_1.4.1 BPCells_0.3.0
[13] Seurat_5.1.0 SeuratObject_5.0.2
[15] sp_2.1-4 harmony_1.2.1
[17] Rcpp_1.0.13 patchwork_1.3.0
[19] lubridate_1.9.3 forcats_1.0.0
[21] stringr_1.5.1 dplyr_1.1.4
[23] purrr_1.0.2 readr_2.1.5
[25] tidyr_1.3.1 tibble_3.2.1
[27] ggplot2_3.5.1 tidyverse_2.0.0
[29] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] fs_1.6.4 spatstat.sparse_3.1-0
[3] httr_1.4.7 RColorBrewer_1.1-3
[5] tools_4.4.1 sctransform_0.4.1
[7] alabaster.base_1.6.1 utf8_1.2.4
[9] R6_2.5.1 HDF5Array_1.34.0
[11] lazyeval_0.2.2 uwot_0.2.2
[13] rhdf5filters_1.18.0 withr_3.0.2
[15] gridExtra_2.3 progressr_0.15.0
[17] cli_3.6.3 spatstat.explore_3.3-3
[19] fastDummies_1.7.4 labeling_0.4.3
[21] alabaster.se_1.6.0 sass_0.4.9
[23] spatstat.data_3.1-2 ggridges_0.5.6
[25] pbapply_1.7-2 parallelly_1.38.0
[27] rstudioapi_0.17.1 RSQLite_2.3.7
[29] generics_0.1.3 ica_1.0-3
[31] spatstat.random_3.3-2 Matrix_1.7-0
[33] fansi_1.0.6 abind_1.4-8
[35] lifecycle_1.0.4 whisker_0.4.1
[37] yaml_2.3.10 rhdf5_2.50.2
[39] SparseArray_1.6.1 BiocFileCache_2.14.0
[41] Rtsne_0.17 grid_4.4.1
[43] blob_1.2.4 promises_1.3.0
[45] ExperimentHub_2.14.0 crayon_1.5.3
[47] miniUI_0.1.1.1 lattice_0.22-6
[49] beachmat_2.22.0 cowplot_1.1.3
[51] KEGGREST_1.46.0 pillar_1.9.0
[53] knitr_1.48 future.apply_1.11.3
[55] codetools_0.2-20 leiden_0.4.3.1
[57] glue_1.8.0 getPass_0.2-4
[59] spatstat.univar_3.0-1 data.table_1.16.2
[61] vctrs_0.6.5 png_0.1-8
[63] gypsum_1.2.0 spam_2.11-0
[65] gtable_0.3.6 cachem_1.1.0
[67] xfun_0.48 S4Arrays_1.6.0
[69] mime_0.12 survival_3.6-4
[71] fitdistrplus_1.2-1 ROCR_1.0-11
[73] nlme_3.1-164 bit64_4.5.2
[75] alabaster.ranges_1.6.0 filelock_1.0.3
[77] RcppAnnoy_0.0.22 rprojroot_2.0.4
[79] bslib_0.8.0 irlba_2.3.5.1
[81] KernSmooth_2.23-24 colorspace_2.1-1
[83] DBI_1.2.3 tidyselect_1.2.1
[85] processx_3.8.4 bit_4.5.0
[87] compiler_4.4.1 curl_5.2.3
[89] git2r_0.35.0 httr2_1.0.5
[91] BiocNeighbors_2.0.1 hdf5r_1.3.11
[93] DelayedArray_0.32.0 plotly_4.10.4
[95] scales_1.3.0 lmtest_0.9-40
[97] callr_3.7.6 rappdirs_0.3.3
[99] digest_0.6.37 goftest_1.2-3
[101] spatstat.utils_3.1-0 alabaster.matrix_1.6.1
[103] rmarkdown_2.28 RhpcBLASctl_0.23-42
[105] XVector_0.46.0 htmltools_0.5.8.1
[107] pkgconfig_2.0.3 sparseMatrixStats_1.18.0
[109] highr_0.11 dbplyr_2.5.0
[111] fastmap_1.2.0 rlang_1.1.4
[113] htmlwidgets_1.6.4 UCSC.utils_1.2.0
[115] shiny_1.9.1 DelayedMatrixStats_1.28.1
[117] farver_2.1.2 jquerylib_0.1.4
[119] zoo_1.8-12 jsonlite_1.8.9
[121] BiocParallel_1.40.0 BiocSingular_1.22.0
[123] magrittr_2.0.3 GenomeInfoDbData_1.2.13
[125] dotCall64_1.2 Rhdf5lib_1.28.0
[127] munsell_0.5.1 reticulate_1.39.0
[129] stringi_1.8.4 alabaster.schemas_1.6.0
[131] zlibbioc_1.52.0 MASS_7.3-60.2
[133] AnnotationHub_3.14.0 plyr_1.8.9
[135] parallel_4.4.1 listenv_0.9.1
[137] ggrepel_0.9.6 deldir_2.0-4
[139] Biostrings_2.74.1 splines_4.4.1
[141] tensor_1.5 hms_1.1.3
[143] ps_1.8.1 igraph_2.1.1
[145] spatstat.geom_3.3-3 RcppHNSW_0.6.0
[147] reshape2_1.4.4 ScaledMatrix_1.14.0
[149] BiocVersion_3.20.0 evaluate_1.0.1
[151] BiocManager_1.30.25 tzdb_0.4.0
[153] httpuv_1.6.15 RANN_2.6.2
[155] polyclip_1.10-7 future_1.34.0
[157] scattermore_1.2 rsvd_1.0.5
[159] xtable_1.8-4 RSpectra_0.16-2
[161] later_1.3.2 viridisLite_0.4.2
[163] memoise_2.0.1 AnnotationDbi_1.68.0
[165] cluster_2.1.6 timechange_0.3.0
[167] globals_0.16.3