Saving Seurat objects
2025-04-16
Last updated: 2025-04-16
Checks: 7 0
Knit directory: muse/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200712) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 41cf543. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Ignored: data/1M_neurons_filtered_gene_bc_matrices_h5.h5
Ignored: data/293t/
Ignored: data/293t_3t3_filtered_gene_bc_matrices.tar.gz
Ignored: data/293t_filtered_gene_bc_matrices.tar.gz
Ignored: data/5k_Human_Donor1_PBMC_3p_gem-x_5k_Human_Donor1_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/5k_Human_Donor2_PBMC_3p_gem-x_5k_Human_Donor2_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/5k_Human_Donor3_PBMC_3p_gem-x_5k_Human_Donor3_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/5k_Human_Donor4_PBMC_3p_gem-x_5k_Human_Donor4_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5
Ignored: data/97516b79-8d08-46a6-b329-5d0a25b0be98.h5ad
Ignored: data/Parent_SC3v3_Human_Glioblastoma_filtered_feature_bc_matrix.tar.gz
Ignored: data/brain_counts/
Ignored: data/cl.obo
Ignored: data/cl.owl
Ignored: data/jurkat/
Ignored: data/jurkat:293t_50:50_filtered_gene_bc_matrices.tar.gz
Ignored: data/jurkat_293t/
Ignored: data/jurkat_filtered_gene_bc_matrices.tar.gz
Ignored: data/pbmc20k/
Ignored: data/pbmc20k_seurat/
Ignored: data/pbmc3k/
Ignored: data/pbmc3k_bpcells_mat/
Ignored: data/pbmc3k_seurat.rds
Ignored: data/pbmc4k_filtered_gene_bc_matrices.tar.gz
Ignored: data/pbmc_1k_v3_filtered_feature_bc_matrix.h5
Ignored: data/pbmc_1k_v3_raw_feature_bc_matrix.h5
Ignored: data/refdata-gex-GRCh38-2020-A.tar.gz
Ignored: data/seurat_1m_neuron.rds
Ignored: data/t_3k_filtered_gene_bc_matrices.tar.gz
Ignored: r_packages_4.4.1/
Untracked files:
Untracked: analysis/bioc_scrnaseq.Rmd
Untracked: pbmc3k_before_filtering.rds
Untracked: pbmc3k_save_rds.rds
Untracked: rsem.merged.gene_counts.tsv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/seurat_save.Rmd) and HTML
(docs/seurat_save.html) files. If you’ve configured a
remote Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 41cf543 | Dave Tang | 2025-04-16 | Save as separate RDS instead of additional assay when using BPCells |
| html | 1de8ba4 | Dave Tang | 2025-04-16 | Build site. |
| Rmd | 445cb01 | Dave Tang | 2025-04-16 | Save additional RDS |
| html | 68b9532 | Dave Tang | 2025-04-16 | Build site. |
| Rmd | e923354 | Dave Tang | 2025-04-16 | Assay classes |
| html | 24107a2 | Dave Tang | 2025-04-16 | Build site. |
| Rmd | 5dd72fd | Dave Tang | 2025-04-16 | Save additional assay |
| html | a22d5e1 | Dave Tang | 2025-04-15 | Build site. |
| Rmd | 77f2810 | Dave Tang | 2025-04-15 | Add miscellaneous data |
| html | 478564c | Dave Tang | 2025-04-15 | Build site. |
| Rmd | deec653 | Dave Tang | 2025-04-15 | Saving Seurat objects |
if ("BPCells" %in% row.names(installed.packages()) == FALSE){
remotes::install_github("bnprks/BPCells/r")
}suppressPackageStartupMessages(library(BPCells))
suppressPackageStartupMessages(library(Seurat))Load Data
Load from my server.
pbmc3k <- readRDS(url("https://davetang.org/file/pbmc3k_seurat.rds", "rb"))
pbmc3kAn object of class Seurat
32738 features across 2700 samples within 1 assay
Active assay: RNA (32738 features, 0 variable features)
1 layer present: countsUse BPCells
Sparse matrix.
class(pbmc3k@assays$RNA$counts)[1] "dgCMatrix"
attr(,"package")
[1] "Matrix"Write a matrix directory and load the matrix using {BPCells}.
my_outdir <- "data/pbmc3k_bpcells_mat"
if(!dir.exists(my_outdir)){
BPCells::write_matrix_dir(
mat = pbmc3k@assays$RNA$counts,
dir = my_outdir
)
}
# Now that we have the matrix on disk, we can load it
pbmc3k.mat <- open_matrix_dir(dir = my_outdir)
pbmc3k.mat32738 x 2700 IterableMatrix object with class MatrixDir
Row names: MIR1302-10, FAM138A ... AC002321.1
Col names: AAACATACAACCAC-1, AAACATTGAGCTAC-1 ... TTTGCATGCCTCAC-1
Data type: double
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc3k_bpcells_matCreate a Seurat object.
pbmc3k_bpcells <- CreateSeuratObject(
counts = pbmc3k.mat,
project = 'pbmc3k',
min.cells = 3,
min.features = 200
)
pbmc3k_bpcells@assays$RNA$counts13714 x 2700 IterableMatrix object with class RenameDims
Row names: AL627309.1, AP006222.2 ... SRSF10.1
Col names: AAACATACAACCAC-1, AAACATTGAGCTAC-1 ... TTTGCATGCCTCAC-1
Data type: double
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc3k_bpcells_mat
2. Select rows: 6, 9 ... 32733 and cols: 1, 2 ... 2700
3. Reset dimnamesSeurat version 4
Mitochondrial percent.
mito.genes <- grep(pattern = "^MT-", x = rownames(x = pbmc3k_bpcells@assays$RNA), ignore.case = TRUE, value = TRUE)
pbmc3k_bpcells[["percent.mt"]] <- PercentageFeatureSet(pbmc3k_bpcells, features = mito.genes)
VlnPlot(pbmc3k_bpcells, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3, layer = "counts")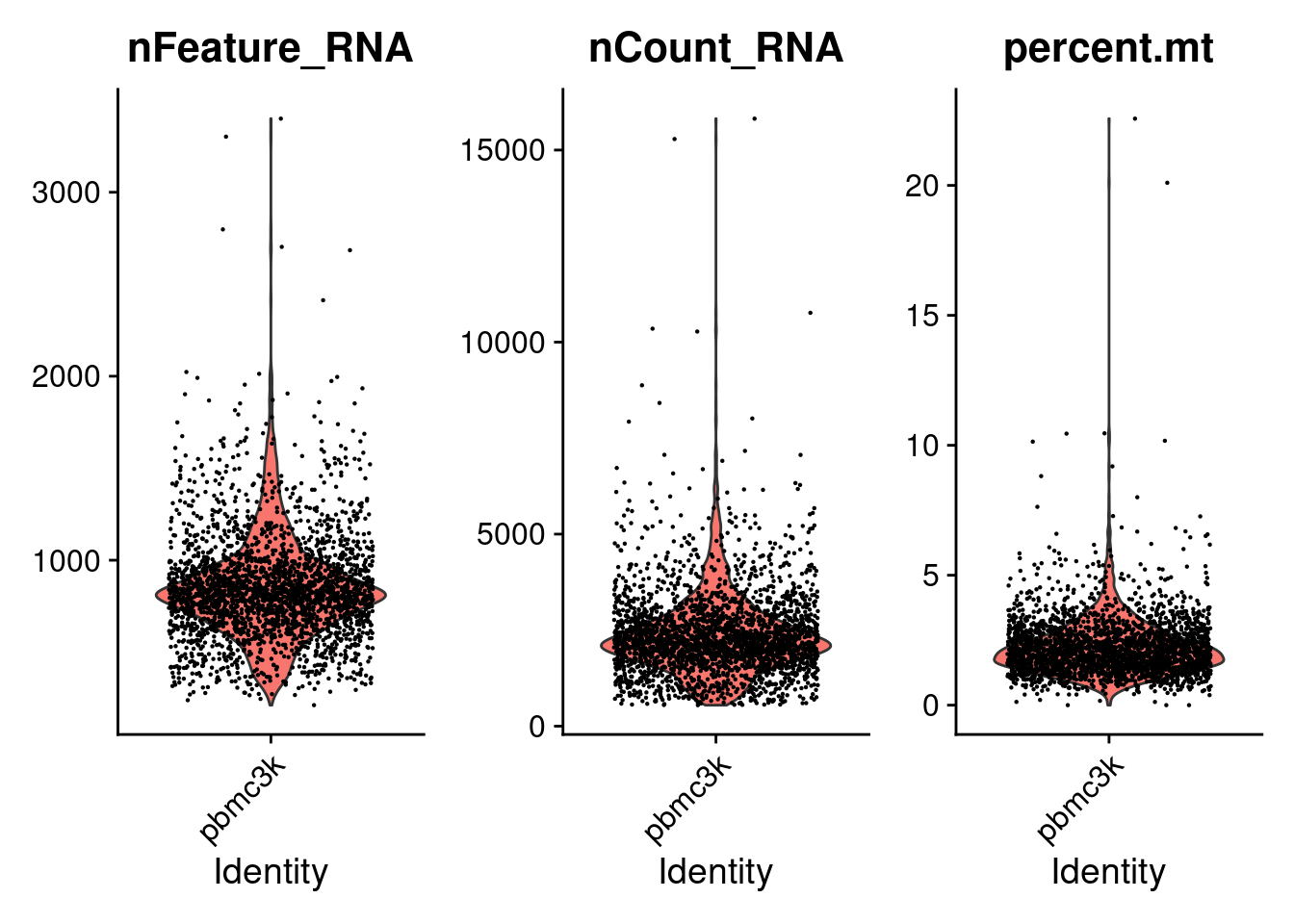
| Version | Author | Date |
|---|---|---|
| 24107a2 | Dave Tang | 2025-04-16 |
Save original data into RAW assay before filtering.
pbmc3k_bpcells[["RAW"]] <- pbmc3k_bpcells@assays$RNAWarning: Key 'rna_' taken, using 'raw_' insteadSeurat::Assays(pbmc3k_bpcells)[1] "RNA" "RAW"Seurat::DefaultAssay(pbmc3k_bpcells)[1] "RNA"Save separate RDS file.
saveRDS(object = pbmc3k_bpcells, file = "pbmc3k_before_filtering.rds")Filter.
pbmc3k_bpcells <- subset(pbmc3k_bpcells, subset = percent.mt < 15)
pbmc3k_bpcellsAn object of class Seurat
27428 features across 2698 samples within 2 assays
Active assay: RNA (13714 features, 0 variable features)
1 layer present: counts
1 other assay present: RAWUnfortunately, the RAW assay becomes filtered as
well.
dim(pbmc3k_bpcells@assays$RNA$counts)[1] 13714 2698dim(pbmc3k_bpcells@assays$RAW$counts)[1] 13714 2698Seurat workflow.
debug_flag <- FALSE
start_time <- Sys.time()
pbmc3k_bpcells <- NormalizeData(pbmc3k_bpcells, normalization.method = "LogNormalize")Normalizing layer: countspbmc3k_bpcells <- FindVariableFeatures(pbmc3k_bpcells, selection.method = 'vst', nfeatures = 2000, verbose = debug_flag)
pbmc3k_bpcells <- ScaleData(pbmc3k_bpcells, verbose = debug_flag)
pbmc3k_bpcells <- RunPCA(pbmc3k_bpcells, verbose = debug_flag)
pbmc3k_bpcells <- RunUMAP(pbmc3k_bpcells, dims = 1:30, verbose = debug_flag)Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
This message will be shown once per sessionpbmc3k_bpcells <- FindNeighbors(pbmc3k_bpcells, dims = 1:30, verbose = debug_flag)
pbmc3k_bpcells <- FindClusters(pbmc3k_bpcells, resolution = 0.5, verbose = debug_flag)
pbmc3k_bpcellsAn object of class Seurat
27428 features across 2698 samples within 2 assays
Active assay: RNA (13714 features, 2000 variable features)
3 layers present: counts, data, scale.data
1 other assay present: RAW
2 dimensional reductions calculated: pca, umapend_time <- Sys.time()
end_time - start_timeTime difference of 11.79401 secsCounts.
pbmc3k_bpcells@assays$RNA$counts13714 x 2698 IterableMatrix object with class RenameDims
Row names: AL627309.1, AP006222.2 ... SRSF10.1
Col names: AAACATACAACCAC-1, AAACATTGAGCTAC-1 ... TTTGCATGCCTCAC-1
Data type: double
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc3k_bpcells_mat
2. Select rows: 6, 9 ... 32733 and cols: 1, 2 ... 2700
3. Reset dimnamesData.
pbmc3k_bpcells@assays$RNA$data13714 x 2698 IterableMatrix object with class RenameDims
Row names: AL627309.1, AP006222.2 ... SRSF10.1
Col names: AAACATACAACCAC-1, AAACATTGAGCTAC-1 ... TTTGCATGCCTCAC-1
Data type: double
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc3k_bpcells_mat
2. Select rows: 6, 9 ... 32733 and cols: 1, 2 ... 2700
3. Reset dimnames
4. Scale by 1e+04
5. Scale columns by 0.000413, 0.000204 ... 0.000504
6. Transform log1p
7. Reset dimnamesScale data.
pbmc3k_bpcells@assays$RNA$scale.data2000 x 2698 IterableMatrix object with class RenameDims
Row names: ISG15, CPSF3L ... MT-ND6
Col names: AAACATACAACCAC-1, AAACATTGAGCTAC-1 ... TTTGCATGCCTCAC-1
Data type: double
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc3k_bpcells_mat
2. Select rows: 43, 63 ... 32708 and cols: 1, 2 ... 2700
3. Reset dimnames
4. Scale by 1e+04
5. Scale columns by 0.000413, 0.000204 ... 0.000504
6. Transform log1p
7. Select rows: 514, 139 ... 580 and cols: all
8. Reset dimnames
9. Transform min by row: 5.2, 19.4 ... 2.3
10. Scale rows by 1.95, 0.55 ... 4.4
11. Shift rows by -0.143, -0.645 ... -0.132
12. Select rows: 636, 1273 ... 455 and cols: all
13. Reset dimnamesPlots
DimPlot(pbmc3k_bpcells)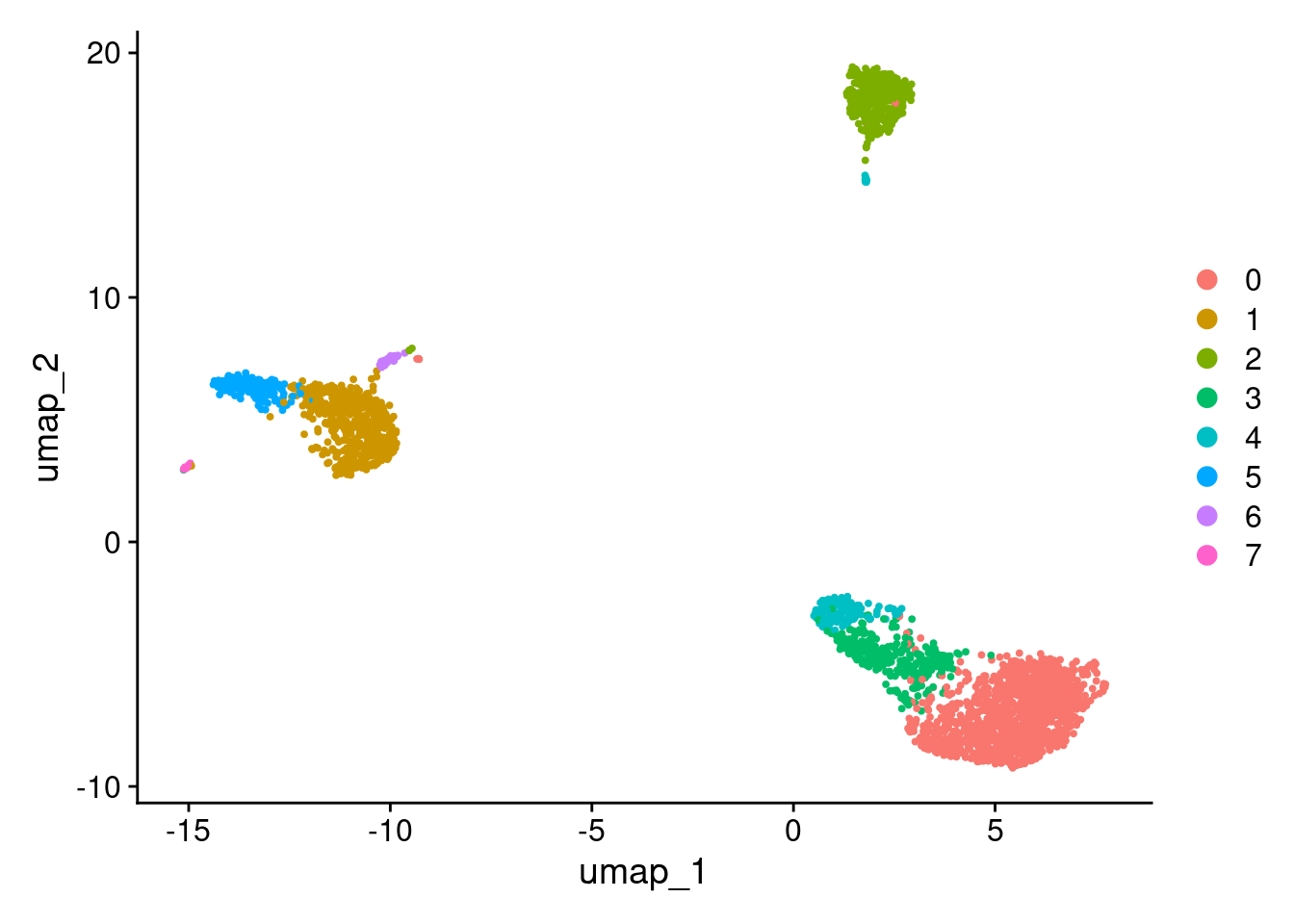
Add miscellaneous data
Get and set miscellaneous data.
Misc(pbmc3k_bpcells)list()Set and output.
Misc(pbmc3k_bpcells, slot = "seed") <- 1984
Misc(pbmc3k_bpcells, slot = "author") <- "Davo"
Misc(pbmc3k_bpcells)$seed
[1] 1984
$author
[1] "Davo"Get specific slot.
Misc(pbmc3k_bpcells, slot = "author")[1] "Davo"Exporting
Save.
saveRDS(object = pbmc3k_bpcells, file = "pbmc3k_save_rds.rds")Load.
pbmc3k_read_rds <- readRDS("pbmc3k_save_rds.rds")
pbmc3k_read_rds@assays$RNA$counts13714 x 2698 IterableMatrix object with class RenameDims
Row names: AL627309.1, AP006222.2 ... SRSF10.1
Col names: AAACATACAACCAC-1, AAACATTGAGCTAC-1 ... TTTGCATGCCTCAC-1
Data type: double
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc3k_bpcells_mat
2. Select rows: 6, 9 ... 32733 and cols: 1, 2 ... 2700
3. Reset dimnamespbmc3k_read_rds@assays$RNA$data13714 x 2698 IterableMatrix object with class RenameDims
Row names: AL627309.1, AP006222.2 ... SRSF10.1
Col names: AAACATACAACCAC-1, AAACATTGAGCTAC-1 ... TTTGCATGCCTCAC-1
Data type: double
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc3k_bpcells_mat
2. Select rows: 6, 9 ... 32733 and cols: 1, 2 ... 2700
3. Reset dimnames
4. Scale by 1e+04
5. Scale columns by 0.000413, 0.000204 ... 0.000504
6. Transform log1p
7. Reset dimnamespbmc3k_read_rds@assays$RNA$scale.data2000 x 2698 IterableMatrix object with class RenameDims
Row names: ISG15, CPSF3L ... MT-ND6
Col names: AAACATACAACCAC-1, AAACATTGAGCTAC-1 ... TTTGCATGCCTCAC-1
Data type: double
Storage order: column major
Queued Operations:
1. Load compressed matrix from directory /home/rstudio/muse/data/pbmc3k_bpcells_mat
2. Select rows: 43, 63 ... 32708 and cols: 1, 2 ... 2700
3. Reset dimnames
4. Scale by 1e+04
5. Scale columns by 0.000413, 0.000204 ... 0.000504
6. Transform log1p
7. Select rows: 514, 139 ... 580 and cols: all
8. Reset dimnames
9. Transform min by row: 5.2, 19.4 ... 2.3
10. Scale rows by 1.95, 0.55 ... 4.4
11. Shift rows by -0.143, -0.645 ... -0.132
12. Select rows: 636, 1273 ... 455 and cols: all
13. Reset dimnamespbmc3k_read_rdsAn object of class Seurat
27428 features across 2698 samples within 2 assays
Active assay: RNA (13714 features, 2000 variable features)
3 layers present: counts, data, scale.data
1 other assay present: RAW
2 dimensional reductions calculated: pca, umapGet miscellaneous data.
Misc(pbmc3k_read_rds)$seed
[1] 1984
$author
[1] "Davo"Check RAW assay.
class(pbmc3k_read_rds@assays$RAW$counts)[1] "RenameDims"
attr(,"package")
[1] "BPCells"class(pbmc3k_read_rds@assays$RNA$counts)[1] "RenameDims"
attr(,"package")
[1] "BPCells"dim(pbmc3k_read_rds@assays$RAW$counts)[1] 13714 2698dim(pbmc3k_read_rds@assays$RNA$counts)[1] 13714 2698Load RDS that was saved before filtering.
pbmc3k_before_filtering <- readRDS("pbmc3k_before_filtering.rds")
class(pbmc3k_before_filtering@assays$RAW$counts)[1] "RenameDims"
attr(,"package")
[1] "BPCells"class(pbmc3k_before_filtering@assays$RNA$counts)[1] "RenameDims"
attr(,"package")
[1] "BPCells"dim(pbmc3k_before_filtering@assays$RAW$counts)[1] 13714 2700dim(pbmc3k_before_filtering@assays$RNA$counts)[1] 13714 2700
sessionInfo()R version 4.4.1 (2024-06-14)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 22.04.5 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] Seurat_5.2.1 SeuratObject_5.0.2 sp_2.2-0 BPCells_0.3.0
[5] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] RColorBrewer_1.1-3 rstudioapi_0.17.1 jsonlite_1.8.9
[4] magrittr_2.0.3 ggbeeswarm_0.7.2 spatstat.utils_3.1-2
[7] farver_2.1.2 rmarkdown_2.28 fs_1.6.4
[10] zlibbioc_1.52.0 vctrs_0.6.5 ROCR_1.0-11
[13] spatstat.explore_3.3-4 htmltools_0.5.8.1 sass_0.4.9
[16] sctransform_0.4.1 parallelly_1.38.0 KernSmooth_2.23-24
[19] bslib_0.8.0 htmlwidgets_1.6.4 ica_1.0-3
[22] plyr_1.8.9 plotly_4.10.4 zoo_1.8-13
[25] cachem_1.1.0 whisker_0.4.1 igraph_2.1.4
[28] mime_0.12 lifecycle_1.0.4 pkgconfig_2.0.3
[31] Matrix_1.7-0 R6_2.5.1 fastmap_1.2.0
[34] GenomeInfoDbData_1.2.13 MatrixGenerics_1.18.1 fitdistrplus_1.2-2
[37] future_1.34.0 shiny_1.10.0 digest_0.6.37
[40] colorspace_2.1-1 patchwork_1.3.0 S4Vectors_0.44.0
[43] ps_1.8.1 rprojroot_2.0.4 tensor_1.5
[46] RSpectra_0.16-2 irlba_2.3.5.1 GenomicRanges_1.58.0
[49] labeling_0.4.3 progressr_0.15.0 spatstat.sparse_3.1-0
[52] polyclip_1.10-7 httr_1.4.7 abind_1.4-8
[55] compiler_4.4.1 withr_3.0.2 fastDummies_1.7.5
[58] highr_0.11 MASS_7.3-60.2 tools_4.4.1
[61] vipor_0.4.7 lmtest_0.9-40 beeswarm_0.4.0
[64] httpuv_1.6.15 future.apply_1.11.3 goftest_1.2-3
[67] glue_1.8.0 callr_3.7.6 nlme_3.1-164
[70] promises_1.3.2 grid_4.4.1 Rtsne_0.17
[73] getPass_0.2-4 cluster_2.1.6 reshape2_1.4.4
[76] generics_0.1.3 gtable_0.3.6 spatstat.data_3.1-4
[79] tidyr_1.3.1 data.table_1.16.2 XVector_0.46.0
[82] BiocGenerics_0.52.0 spatstat.geom_3.3-5 RcppAnnoy_0.0.22
[85] ggrepel_0.9.6 RANN_2.6.2 pillar_1.10.1
[88] stringr_1.5.1 spam_2.11-1 RcppHNSW_0.6.0
[91] later_1.3.2 splines_4.4.1 dplyr_1.1.4
[94] lattice_0.22-6 deldir_2.0-4 survival_3.6-4
[97] tidyselect_1.2.1 miniUI_0.1.1.1 pbapply_1.7-2
[100] knitr_1.48 git2r_0.35.0 gridExtra_2.3
[103] IRanges_2.40.1 scattermore_1.2 stats4_4.4.1
[106] xfun_0.48 matrixStats_1.5.0 stringi_1.8.4
[109] UCSC.utils_1.2.0 lazyeval_0.2.2 yaml_2.3.10
[112] evaluate_1.0.1 codetools_0.2-20 tibble_3.2.1
[115] cli_3.6.3 uwot_0.2.3 xtable_1.8-4
[118] reticulate_1.41.0 munsell_0.5.1 processx_3.8.4
[121] jquerylib_0.1.4 Rcpp_1.0.13 GenomeInfoDb_1.42.3
[124] spatstat.random_3.3-2 globals_0.16.3 png_0.1-8
[127] ggrastr_1.0.2 spatstat.univar_3.1-2 parallel_4.4.1
[130] ggplot2_3.5.1 dotCall64_1.2 listenv_0.9.1
[133] viridisLite_0.4.2 scales_1.3.0 ggridges_0.5.6
[136] purrr_1.0.2 rlang_1.1.4 cowplot_1.1.3