Workflow management system
Dave Tang
2026-01-07
Last updated: 2026-01-07
Checks: 7 0
Knit directory: bioinformatics_tips/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200503) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 60b432b. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/pipelining.Rmd) and HTML
(docs/pipelining.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 97b7d02 | Dave Tang | 2023-06-27 | Build site. |
| html | 8a0e171 | davetang | 2020-12-15 | Build site. |
| Rmd | 176d41e | davetang | 2020-12-15 | Include figures |
| html | 75339f4 | davetang | 2020-12-15 | Build site. |
| Rmd | 957ab5e | davetang | 2020-12-15 | Elaborate on WDL and Cromwell |
| html | 0be4b55 | davetang | 2020-08-23 | Build site. |
| Rmd | f52afb5 | davetang | 2020-08-23 | Update |
| html | 3e6869c | davetang | 2020-08-15 | Build site. |
| Rmd | 93ad1d0 | davetang | 2020-08-15 | Update |
| html | e9934e7 | davetang | 2020-06-07 | Security basics |
| html | b45e21b | davetang | 2020-05-23 | Build site. |
| Rmd | 3a4d719 | davetang | 2020-05-23 | Workflow management system |
A workflow management system (WMS) is software that makes it easier to implement, execute, and manage workflows. If you work with high-throughput sequencing (HTS) data, chances are that you will benefit from using a WMS. A typical HTS workflow involves various processing steps that run sequentially, for example in RNA sequencing (RNA-seq) the raw data is first analysed using quality control (QC) tools and filtered to remove data that is “bad” quality. This filtered dataset is then aligned to a reference sequence and the gene/transcript abundance is calculated. Once you have set up the workflow using a WMS, you can simply run the workflow for new data saving you time and also reducing the potential for human error and increasing reproducibility.
Initially, it may take a bit more time to implement your workflow under a WMS and you may be familiar with the following comic, where in practice automating a task is much more difficult than in theory.

However in my experience, once you learn the basics of a WMS it becomes easier and easier to implement your workflows under such a system and I do end up freeing up my time to do other tasks as per this tweet.
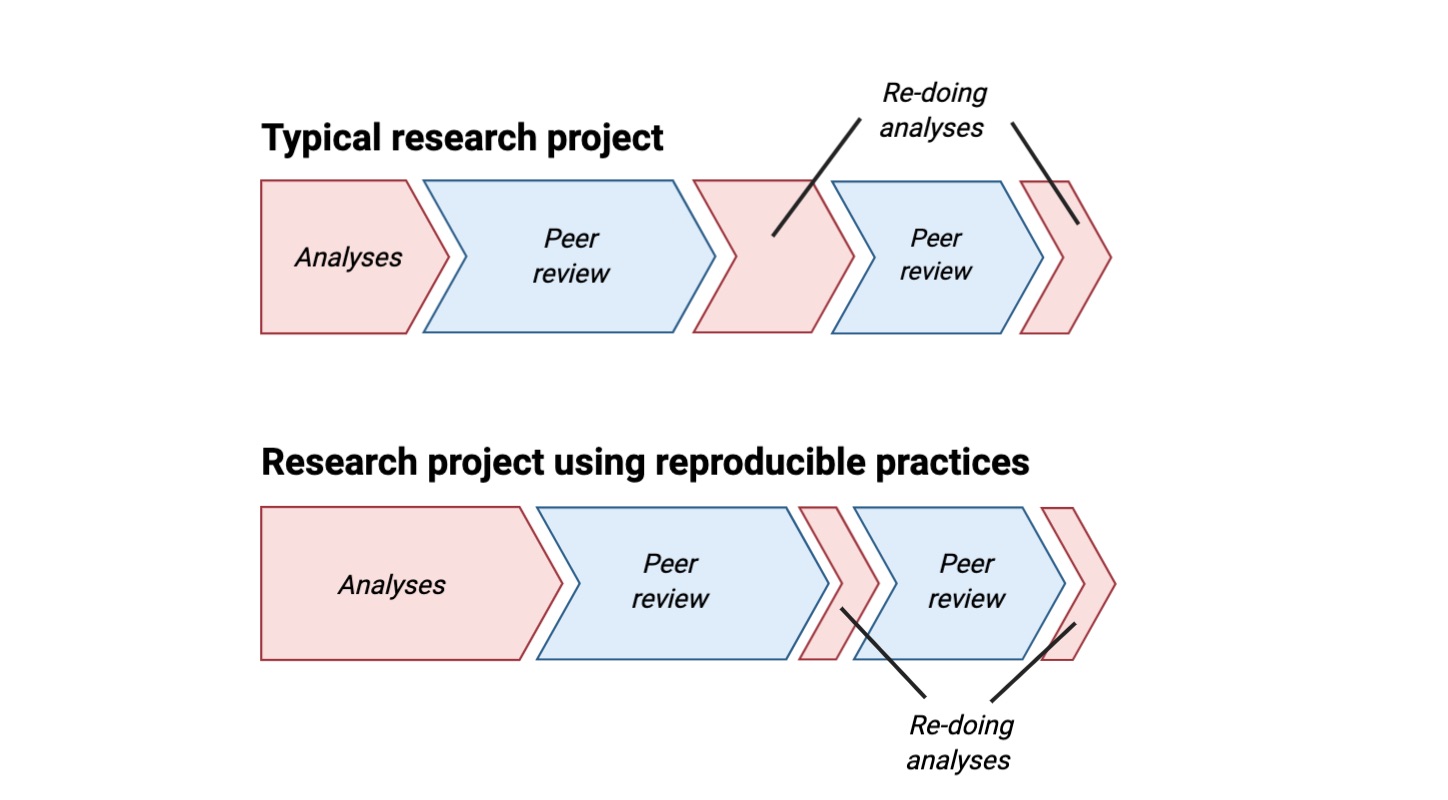
While it is entirely possible to implement workflows using simply scripts written in shell (or some other scripting language) WMSs offer many additional features besides running your workflow. For example, workflows implemented in Workflow Description Language (WDL) can be easily executed across different platforms, such as locally or on a High Performance Computing (HPC) cluster, by using Cromwell. A really useful of Cromwell is called “Call Caching” and can be used to resume a job if it failed halfway through execution and use previously computed data. For example, if you wanted to test a new gene quantification tool for your RNA-seq workflow but use the same approach for the QC and read mapping, call caching will copy (or link) your previously mapped data and only run the new gene quantification tool. WDL has always been advertised as a Domain-Specific Language (DSL) that is easy to read and write regardless of your computational background. If I have piqued your interest, you can check out my blog post on learning WDL.
Workflow management systems
Besides WDL, there are other workflow management systems that include Snakemake, Bpipe and Nextflow, which both are based on Groovy. A survey conducted on Twitter has a list of other systems and showed that Snakemake is the most popular. There is a nice discussion on Reddit on the strengths and weaknesses of different WMSs.
Another advantage of using a WMS is that it is very likely that your workflow of interest has already been implemented. Below are some repositories that contain various workflows implemented in their respective WMS:
- Snakemake - https://github.com/snakemake-workflows
- Nextflow - https://github.com/nextflow-io/awesome-nextflow
- WDL - https://github.com/biowdl and https://github.com/gatk-workflows
Even if the available workflows do not exactly match your specifications, you can find a similar pipeline and modify it as you wish.
Example
In the WDL, you set up each component of your workflow as individual tasks, which follows a defined structure:
task hello {
input {
String pattern
File in
}
command {
egrep '${pattern}' '${in}'
}
runtime {
docker: "ubuntu:latest"
memory: "4G"
cpu: "3"
}
output {
Array[String] matches = read_lines(stdout())
}
}A task is like a function and takes an input or inputs and processes the input according to the command block and generates an output or outputs. In addition, you can also set up specify resource usage per task, such as how much memory and CPU can be used. While the Broad team recommend the use of Docker, it is not strictly necessary and tools can be executed via other means. After defining all your tasks, the workflow is created by calling all your tasks and specifying the inputs of each task; task dependencies are created by specifying the output of one task as the input for another task or tasks.
A JSON file is used as a configuration file for your workflow and will specify parameters and the location of files, such as your raw input and reference files. Finally, the execution engine called Cromwell is used to execute the workflow and handle all the logging, resource management, and pipeline execution.
Snakemake on the other hand uses rules to set up each part of your workflow and they are similar to tasks in WDL.
rule sort:
input:
"test.txt"
output:
"test.sorted.txt"
shell:
"sort -n {input} > {output}"If we run the example above using Snakemake, the input file
test.txt will get sorted numerically and the output is
stored in test.sorted.txt. Typically, you would write a
pipeline (a Snakefile) that takes input from a config file
(e.g. config.yaml). If you wanted to run the pipeline for a
new dataset, you will just need to create a new config file. I have a short
blog post on Snakemake too.
WDL
For a nice introduction to WDL and Cromwell, listen to Getting started with WDL and Cromwell presented by Ruchi Munshi. A quick summary of the talk:
- WDL and Cromwell spun out from the need of a new tool and platform that can be easily scaled to process a lot of data
- WDL is aimed at being a DSL that is easy for humans to read and write, in particular for biomedical scientists
- Workflows implemented in WDL can be executed using Cromwell at any scale across different platforms, such as your local computer, on HPC clusters, and on various cloud platforms.
- The use of Docker containers is recommended for portability and reproducibility
- WDL and Cromwell have been used to process a lot of data at the Broad Institute, which goes to show that it is a well supported tool that is under constant development
Some links to check out:
- https://github.com/FredHutch/reproducible-workflows
- https://sciwiki.fredhutch.org/compdemos/Cromwell/
- Specification - https://github.com/openwdl/wdl/tree/main/versions
- Optional inputs - https://github.com/openwdl/wdl/blob/master/versions/1.0/SPEC.md#optional-inputs
- true and false - https://github.com/openwdl/wdl/blob/master/versions/1.0/SPEC.md#true-and-false
sessionInfo()R version 4.5.0 (2025-04-11)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 24.04.3 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] vctrs_0.6.5 httr_1.4.7 cli_3.6.5 knitr_1.50
[5] rlang_1.1.6 xfun_0.52 stringi_1.8.7 processx_3.8.6
[9] promises_1.3.3 jsonlite_2.0.0 glue_1.8.0 rprojroot_2.0.4
[13] git2r_0.36.2 htmltools_0.5.8.1 httpuv_1.6.16 ps_1.9.1
[17] sass_0.4.10 rmarkdown_2.29 jquerylib_0.1.4 tibble_3.3.0
[21] evaluate_1.0.3 fastmap_1.2.0 yaml_2.3.10 lifecycle_1.0.4
[25] whisker_0.4.1 stringr_1.5.1 compiler_4.5.0 fs_1.6.6
[29] pkgconfig_2.0.3 Rcpp_1.0.14 rstudioapi_0.17.1 later_1.4.2
[33] digest_0.6.37 R6_2.6.1 pillar_1.11.0 callr_3.7.6
[37] magrittr_2.0.3 bslib_0.9.0 tools_4.5.0 cachem_1.1.0
[41] getPass_0.2-4